
Podcast: Play in new window | Download (Duration: 6:24 — 9.1MB)
Subscribe: Apple Podcasts | Spotify | RSS
Welcome back to the tasty morsels of critical care podcast.
Today we’re going to talk about some of the basics of some of our favorite drugs intensive care – the diuretics. As always this is planned to be a brief overview of the essentials rather than the deep dive.
As a starter pretty much all diuresis is conducted by convincing the kidney to lose more Na. Lose the Na and the water will follow.
First on the list of course is furosemide. This is one of the commonest drugs we use in intensive care and we really should just be mixing it in with the NG feed or the propofol given how commonly we use it.
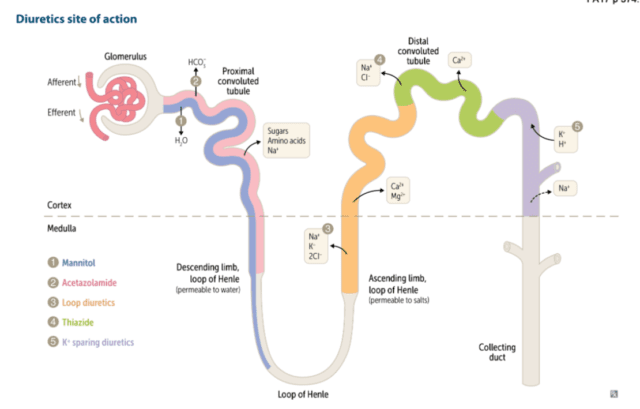
Furosemide is one of several loop diuretics. By loop we mean its site of action is at the loop of henle, site of the much loved countercurrent mulitplication system. In particular furosemide acts by blocking the NaK2Cl pump in the thick ascending loop of henle. This sounds all very technical and impressive but how does blocking said channel cause an increase in the wee wee in the bag? Ultimately you end up with a lot more sodium arriving at the collecting duct. The presence of extra sodium in the collecting duct decreases the osmotic gradient between the medulla and the tubule and as a result less water is reabsorbed and more comes out in the urine.
Furosemide is normally highly protein bound. As a result it can’t get into the nephron through the glomerulus, which in the healthy state won’t let large things like albumin through. Therefore to get to its site of action in the loop of Henle it gets secreted into the proximal tubule and washed along with the ultrafiltrate towards the loop. This feature of secretion in the proximal tubules is one of the things we see with the furosemide stress test, typically used to predict need for RRT in AKI. A lack of response to a healthy (ie at least 1mg/kg) dose of furosemide tells us the proximal tubules are in big trouble and there will be a likely need for RRT.
In terms of side effects the ones that are perhaps clinically most apparent are the electrolyte losses (primarily potassium and magnesium) and hypernatraemia (as the water loss is in excess of the Na loss). There is a corresponding “contraction alkalosis” that is nicely explained at the deranged physiology post or in audio form over at the curious clinicians. Longer term the one worth knowing about is the ototoxicity commonly seen with high doses of furosemide especially in conjunction with our other favourite ototoxic drugs – the aminoglycosides. Though to be honest, only our most chronically critically ill patients stay long enough with us for us to pick up the ototoxicity.
Next on the list are our thiazides. Typically for our local practice that means metolazone. Thiazides work just a little further down the windy nephron river from the loop of Henle at the distal convoluted tubule. Once there it inhibits the NaCl transporter system again meaning increased delivery of Na to the distal tubule where most of the water reabsorption occurs. Thiazides aren’t especially powerful as a diuretic strategy but they are additive from a Na wasting (and hence water losing) perspective as you’re targeting a different part of the nephron. I find the metolazone often gets added when you still want to diurese but you’re a bit worried about the rising Na. The idea is that you get the Na content of the urine to the sweet spot where you lose equal amounts of salt and water and the serum concentration stays the same. Like most things in ICU this is likely physiological wishful thinking rather than good science and it keeps us amused while the disease process resolves on its own.
Continuing our journey through the nephron we have the aldosterone receptor antagonists. A class largely occupied by spironolactone. Spiro (to its friends) works by blocking the very important ENaC (or epithelial sodium channel), especially in the collecting duct. When these are blocked Na no longer is reabsorbed in the collecting duct and hey presto water follows the Na out of the nephron into the ureters. The main side effect of the increased concentration of Na in the collecting duct will be a reluctance to secrete K into the duct thus preventing K wastage and ultimately increasing the serum K. It is not an especially effective diuretic in terms of producing volumes of urine but more importantly it does have a significant long term mortality benefit in patients with heart failure unlike crowd favourite furosemide. It is of course difficult to extrapolate findings from massive cardiology heart failure trials to the ventilated patient with a dodgy ticker with multi organ failure in the ICU but there you go.
The final drug we’ll mention today is acetazolamide. It has its site of action way back in the early nephron at the proximal convoluted tubule. It is a carbonic anhydrase inhibtor, unsurprisingly inhibiting the action of carbonic anhydrase. From the name “carbonic anydrase” we can hopefully deduce that it inhibits the process of removing water from carbonic acid. Ultimately this impairs HCO3 reabsorption at the proximal tubule creating a scenario somewhat similar to renal tubular acidosis. The drug clearly causes a diuresis and does indeed increase the Na wasted in the urine though the precise mechanism is not entirely clear. My anecdotal experience when taking the stuff climbing Kilimanjaro nearly 20 years ago suggests indeed it does make you want to pee a lot more. There are a a few small trials looking at its use in ICU none of which are hugely compelling for benefit but I find myself reaching for it when the fursoemide driven alkalosis is causing issues or you’re playing a game of “diuresis jedi” and want to complete all the steps of the “nephron bomb”
Reading:
Deranged Physiology
Mullens, W., Verbrugge, F. H., Nijst, P. & Tang, W. H. W. Renal sodium avidity in heart failure: from pathophysiology to treatment strategies. Eur Heart J 38, 1872–1882 (2017).